来源:先进能源材料化学教育部重点实验室
JACS:双极水系电解液的设计

可充电水系电池由于其高安全性和低成本,使其作为一种潜在的大规模能源存储系统。然而,开发具有高可持续性和可逆性的水系电池是具有挑战性的。
成果简介
在此,南开大学陈军院士等人报道了一种两性羟基乙酸铝(AlAc(OH)2)电解液,其具有H+和OH−的双极电解化能力,有助于蒽醌(AQ)负极和氢氧化镍(Ni(OH)2)正极的氧化还原反应。其中,AlAc(OH)2(H2O)3溶剂化结构的双极电离能力是Al3+和OH−的强极化能力的结果。在解离常数为5.0/3.0时,H+/OH−的解离能力强于水(14.0),提高了电极的氧化还原反应。具体来说,H+的吸收阻止AQ负极离子键的形成,抑制电极溶解,而OH−为Ni(OH)2正极的稳定转化反应提供了局部碱性环境。进一步验证,设计的AQ||Ni(OH)2电池的AQ负极放电容量为243.9 mAhg-1,循环300次容量保持率为78.2%。此外,作者组装了一个放电容量为0.90 Ah的软包电池,基于电池的总质量展现出44.7 Whkg-1的能量密度。这项工作显著地拓宽了水系电池的类型,并代表了双极电解液的设计理念和与H+和OH−进行的独特的电化学反应。
相关文章以“Sustainable Aqueous Batteries Based on Bipolar Dissociation of Aluminum Hydroxyacetate Electrolyte”为题发表在J. Am. Chem. Soc.上。
研究背景
可再生能源的高效利用和转换需要大规模的储能系统。能量密度高、循环寿命长的锂离子电池已广泛应用于便携式电子设备和电动汽车中。然而,锂资源短缺、成本高、生产环境恶劣、有机电解液的可燃性和安全问题不可避免地制约了锂离子电池在大规模储能中的应用。相比之下,基于水系电解液的电池具有安全、环保、组装环境要求低等优点,被认为是大规模储能系统的有前途的候选者。因此,开发高性能水系电池具有重要意义。
水系电池的发展可以追溯到1800年,当时Volta通过堆叠锌箔、电解液和铜箔发明了第一个电池(Volta pile)。后来,基于Zn负极和MnO2正极的干电池和碱性电池得到进一步发展。然而,这种原电池很难可逆地充电和放电。虽然商业化铅酸电池是低成本的二次电池,但它们仍然受到循环寿命短、能量密度低、铅对环境污染等限制。基于Ni(OH)2的镍镉和镍氢(Ni-MH)电池具有良好的充放电性能,但负极存在成本高且潜在的析氢反应(HER)。开发不含HER的低成本负极材料对于具有高性能Ni(OH)2的全电池是必要的。有机醌基材料具有可持续性、高可逆性和可调电位等优点,使其适用于负极材料。但醌的还原产物会解离,易溶于碱性电解液,导致容量衰减,循环性能差。此外,广泛使用的强酸性或碱性电解液具有很强的腐蚀性,不适合大规模应用。因此,开发具有双极电离能力的中性电解液对于支持下一代醌-Ni(OH)2的开发是必要的,且具有挑战性。
图文导读
H+/OH−化学中性水系电解液的设计策略
虽然传统的电池体系利用H+和OH−反应具有良好的电化学性能,他们通常需要特殊的电池结构来适应强酸性和碱性电解液和阴离子/阳离子交换膜,这是昂贵的且与性能电池组装水平密切相关。同时,这些膜的选择性不是100%,导致长时间运行后产生穿梭效应,导致电池故障。相比之下,中性电解液含量适中,具有大规模应用的潜力。由于Al3+电荷高,离子半径小,具有很强的极化能力,可以削弱水合Al3+溶剂化结构中水分子的O-H键,从而激活H+的电离能力。因此,本文选择了两性铝盐AlAc(OH)2来构成一种既能同时电离H+又能电离OH−的电解液。H+用于促进有机负极醌-苯酚转化,而不是形成蒽醌类盐与金属负离子结合,从而导致电极溶解。OH−负离子协助和支持Ni(OH)2正极在充电-放电过程中的转换反应。基于Ni(OH)2正极和AQ负极,电极反应机理如图1所示,方程如下:
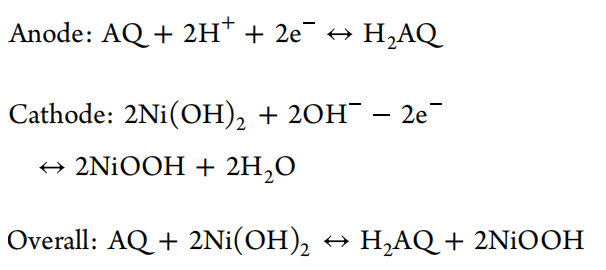
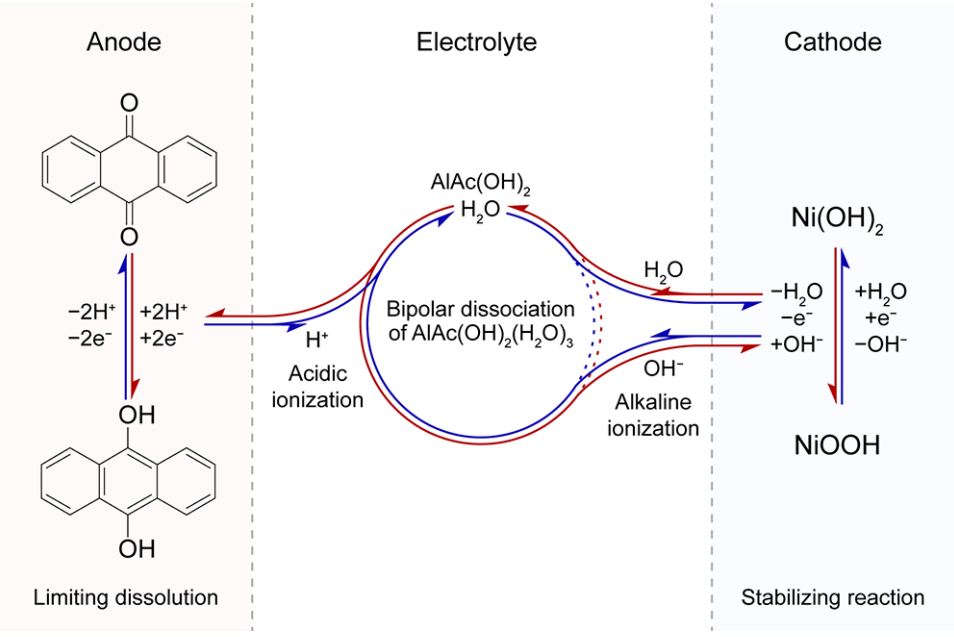
图1. 设计了含AlAc(OH)2电解液、AQ负极和Ni(OH)2正极的水系电池系统
电解液结构与H+/OH−电离机理
同时,采用DFT计算了AlAc(OH)2电解液溶剂化结构的H+/OH−解离常数(pKa/pKb)。为了明确地考虑溶剂效应,添加了12个水分子作为AlAc(OH)2(H2O)3的第二溶剂化壳层。它的酸性电离产生H+和[AlAc(OH)3(H2O)14]-,pKa的解离常数为5.0(图2a)。当它经过碱性电离时,OH−和[AlAc(OH)(H2O)15]+的pKb为3.0,如此低的解离常数表明了双极电离的可行性。传统上,铝盐溶液中的Al3+溶剂化结构被认为是Al(H2O)63+,由此生成H+和[Al(OH)(H2O)17]2+时的pKa为4.8,略低于[AlAc(OH)(H2O)15]+的pKa。由于[Al(H2O)18]3+不含OH−,因此OH−的电离只能由[Al(OH)(H2O)17]2+的碱性电离产生,计算得到的pKb为11.0(图2b)。

图2. AlAc(OH)2水系电解液的H+/OH−电离机理和电解液结构
电解液与电极之间的相容性
由于H+和OH−具有较强的电离能力,利用AlAc(OH)2电解液分别研究了H+和OH−涉及AQ负极和Ni(OH)2正极在三电极电池中的氧化还原反应。与Ni(OH)2正极和AQ负极相结合的电池具有潜在的适用性,在电池系统中具有最高的能量密度。由于AlAc(OH)2电解液可以发生碱性电离,因此产生的OH−阴离子可以协助Ni(OH)2正极在充/放电过程中的转化反应。因此,Ni(OH)2正极在AlAc(OH)2电解液中的库仑效率高于传统的铝电解液,如硫酸铝和AlAc3电解液(图3a)。同时,AlAc(OH)2电解液增强了Ni(OH)2的循环性能(图3b),这是局部碱性环境的结果。此外,采用原位紫外光谱(UV−vis)光谱法揭示了AQ负极的溶解情况。当与LiAc电解液匹配时,AQ负极会形成蒽喹诺盐,并发生快速溶解(图3c)。

图3. AlAc(OH)2电解液与Ni(OH)2正极/AQ负极的兼容性
AQ负极的氧化还原机理
此外,AQ的氧化还原机制首先通过核磁共振得到验证。在起始状态下,AQ在原苯环上有两种H原子,分别位于8.16(a1)和7.88(b1)ppm。充电后,峰值变为8.34 ppm(a2)和7.42ppm(b2)(图4a)。同时,代表羟基上H原子的峰出现在9.43 ppm(c2)ppm。a2、b2和c2的峰面积之比为2:2:1。放电后,H2AQ完全转化为AQ,表明在AQ负极中H+的可逆摄取/去除。对还原产物的进一步测试表明,AQ的还原过程主要是由质子化反应决定的,而不是Al3+的插入。通过原位XRD实验,揭示了AQ在还原过程中晶体结构的变化。在充电过程中,AQ(200)和(−201)晶面11.32和14.18°的峰逐渐消失,H2AQ 11.36和140.78°代表(010)和(002)的峰出现(图4b)。此外,在两次循环试验中,AQ峰具有良好的重现性,表明在H+吸收/去除后,晶体发生了稳定的结构变化。

图4. AQ负极的电化学氧化还原机制和H+迁移
全电池的电化学性能
为了研究电池系统的实用性,作者组装了一个基于AQ负极、AlAc(OH)2电解液和Ni(OH)2正极的全电池。与Al2(SO4)3(2.16V)电解液和KOH(1.62 V)电解液相比,AlAc(OH)2电解液的电化学窗口更宽(2.35V)(图5a)。Ni(OH)2正极和AQ负极在电压间隙为1.30 V的AlAc(OH)2电解液中表现出高可逆性,且通过原位pH测试观察到全电池运行中H+/OH−的产生和消耗。在充电过程中,正极侧的pH减少,而负极侧的pH增加,这分别是由于两侧H+和OH−的消耗造成的(图4b)。在竖立过程中,随着H+和OH−迅速扩散并在电解液中中和,两侧的pH恢复到初始值。在放电过程中,pH的变化成反比。放电后,pH返回到初始值,证明了H+和OH−的产生和消耗的同时性。此外,原位差分电化学质谱(DEMS)表明,在充/放电过程中,具有宽电化学窗口的电池既不产生H2也不产生O2(图5c)。

图5. AQ|AlAc(OH)2|Ni(OH)2全电池的电化学性能
总结展望
综上所述,本文开发了一种基于两性羟基乙酸铝(AlAc(OH)2)电解液,使蒽醌(AQ)负极和Ni(OH)2正极中H+和OH−参与反应分别具有不同的电化学机制,通过从头算分子动力学(AIMD)和密度泛函理论(DFT)的计算,证明了AlAc(OH)2(H2O)3的溶剂化结构和双极电离机理。同时,通过核磁共振(NMR)、X射线衍射(XRD)、拉曼光谱和DFT计算,验证了具有H+/OH−电极的电化学氧化还原机制。本文所设计的AQ|AlAc(OH)2|Ni(OH)2电池具有低电极溶解,无O2/H2析出且具有高可逆性,循环300次后放电容量为243.9 mAhg-1anode,容量保持率为78.2%。这项工作提出了一种新的双极水系电解液的设计理念,并开发了具有独特的H+/OH−解离机制的电池系统。
文献信息
Qiu Zhang,† Xiaomeng Liu,† Yong Lu,† Youxuan Ni, Weiwei Xie, Zhenhua Yan, Fujun Li,and Jun Chen*, Sustainable Aqueous Batteries Based on Bipolar Dissociation of Aluminum Hydroxyacetate Electrolyte, J. Am. Chem. Soc.. (2024). https://doi.org/10.1021/jacs.3c139632
Angew:温度-压力诱导策略提升ORR性能!

氧还原反应(ORR)在金属空气电池和燃料电池发展中扮演着重要作用,其中应变调节可以改变催化剂的几何形状,调整催化剂的表面电荷分布,是优化ORR活性的有力策略,但在材料中引入应变调节仍然难以实现催化性能的大幅度提升。
在此,南开大学陈军院士和严振华副研究员等人提出了一种温度-压力诱导策略来实现金属配位聚合物的晶格应变调控。通过对应变效应对ORR性能的系统研究,进一步理解和证实了几何效应和电子效应之间的关系,晶格压缩率为2.0%的应变Co-DABDT(DABDT=2,5-二氨基苯-1,4-二硫醇)表现为0.81 V。理论分析表明,晶格应变改变S原子周围的自旋电荷密度,然后调节氢键相互作用与中间体促进ORR催化过程。这项工作有助于从原子水平上理解催化机理。
相关文章以“Lattice Strained Induced Spin Regulation in Co-N/S Coordination Framework Enhanced Oxygen Reduction Reaction”为题发表在Angew. Chem. Int. Ed.上。
研究背景
基于氧还原反应(ORR)的金属-空气电池和燃料电池等,是实现全球碳中和的重要途径。然而,由于ORR的过程复杂,其动力学过程通常被认为是金属-空气电池和燃料电池性能的瓶颈。目前,最有效的商业化ORR催化剂是铂基材料,但其昂贵性限制了其商业应用。追求具有低过电位、高质量活性和高耐久性的ORR电催化剂遇到较大的挑战。金属-有机框架(MOFs)具有明确的结构和丰富的活性金属位点,有效地结合了均相和多相催化剂的优点。然而,对于传统的MOFs,其电化学活性的提高受到电绝缘体的严重抑制,这严重限制了其在电催化中的广泛应用。最近,d-π共轭金属配位框架已经成为一类新的MOF,其平面内π离域和弱平面外π叠加,有效地提高了电子转移能力,且仍然保留了传统MOFs的固有优势。因此,d-π共轭金属配位框架为研究ORR电催化机理提供了一个理想的模型结构。同时,晶格应变可以有效地诱导和调节其几何结构和电子结构,是提高ORR性能的重要方法。然而,应变的产生通常是无法控制的。由于主体和客体之间的相互作用,以及它们与传统无机材料相比增强的灵活性,MOFs在其晶体结构上表现出易变形,使其成为应变诱导化学检测的优秀候选材料。目前,MOFs的调控主要考虑金属位点的配位数、配位原子和轴向客体群。然而,应变诱导的MOFs调控很少,这是目前具有挑战性和讨论。值得注意的是,由晶格应变引起的空间结构的变化可以实现对催化剂的电子态的微观调控,这可能是提高MOFs催化活性的有效策略。
图文导读
以新型d-π共轭Co-DABDT(Co-N2S2)为模型催化剂,对晶格应变进行了微调,并分析了应变效应对ORR电催化性能的影响。在不同温度下,通过络合反应成功地合成了Co-DABDT,其由大量堆叠的纳米棒组成的。为了进一步分析纳米棒的微观形貌,从图1b-e中的TEM可以看出,所有Co-DABDT样品都是均匀的矩形纳米棒结构,长度和宽度均为分别为70 nm和10 nm。根据HRTEM(图1b-e),在50 oC下得到的Co-DABDT的条纹晶格参数为5.89 Å。值得注意的是,当反应体系的温度升高到70、120和180℃时,(100)晶面被压缩成5.77 Å、5.69 Å和5.64 Å,它们对应的晶格压缩比分别为2.0%、3.4%和4.2%。

图1. 通过晶格压缩调节S位点自旋电荷密度的示意图及表征
同时,作者使用粉末x射线衍射(PXRD)进一步证明了Co-DABDT中存在应变效应。根据图2a中原始Co-DABDT的PXRD图谱,位于11.24、15.20和17.74°的尖锐特征峰分别归因于(001)、(100)和(-101),表明其具有高度结晶结构。原始和应变后的Co-DABDT的特征衍射峰与理论XRD模拟一致,证实了高结晶Co-DABDT的合成。此外,(100)平面的衍射角显著移动到15.52,15.76和15.84°,如图2b所示,这也是Co-DABDT晶格应变的重要证据。其结果可能是随着系统温度的升高,系统的环境压力增大,导致Co-DABDT沿[100]方向被压缩(图2c)。利用傅里叶变换红外光谱(FT-IR)验证了Co-DABDT的键合特性的演化。通过FTIR谱比较反应前后的结构变化(图2d),DABDT的S-H伸缩振动位于2500 cm-1,C-N和C-S伸缩振动分别出现在1183和1038 cm-1。、

图2. 理化性质的研究
为了进一步探索晶格应变与ORR催化活性之间的内部关系,作者利用旋转环圆盘电极(RRDE)对合成的Co-DABDT在O2饱和0.1 M氢氧化钾溶液中对ORR性能进行了评价。首先使用线性扫描伏安法来估计催化活性。当电极旋转速率为1600轮每分钟(rpm)和扫描速度为5 mV-1时,Co-DABDT,Co-DABDT-2.0%,Co-DABDT-3.4%和Co-DABDT-4.2%的起始电位分别是0.88 V,0.96 V,0.94 V和0.87V,表明Co-DABDT-2.0%ORR反应有更好的电催化活性。同时,根据极化曲线,可以计算出电催化ORR过程中的电子转移数和过氧化氢产率(图3b)。根据圆盘的电流,在0.3~0.8 V的电压范围内,Co-DABDT、CoDABDT-2.0%、Co-DABDT-3.4%和Co-DABDT-4.2%的平均电子转移数约为3.81、3.85、3.61和3.54(图3c)。
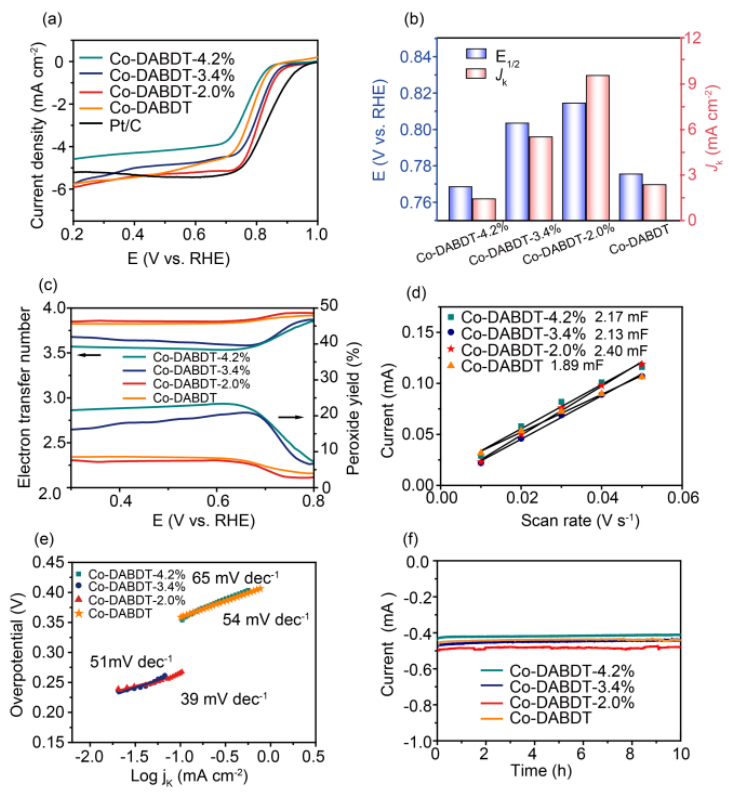
图3. 晶格应变与ORR催化活性之间的内部关系
为了了解应变调节电催化之间的内部关系,作者通过DFT计算进一步分析了电催化剂和ORR过程的电子性质(图4a)。图4b显示了不同压缩应力下Co-DABDT的自旋电荷密度的分布。结果表明,S原子周围的自旋电荷密度先增大后减小。在这些Co-DABDT催化剂中,Co-DABDT-2%在S原子周围的自旋电荷密度最高。当OOH被吸附在Co-DABDT-2%的催化剂上时,OOH的H原子与配位S原子之间出现分子内氢键相互作用,如电荷密度差所示(图4d)。此外,还发现Co-DABDT-2%催化剂的S原子在关键中间体OOH吸附后获得的电子最多,表明S原子和H原子之间的静电相互作用更强(图4c)。因此,Co-DABDT-2%催化剂对关键中间体OOH具有较强的稳定性。不同压缩应力下Co-DABDT催化剂的自由能图如图4e所示。在这三种催化剂中,Co-DABDT-2%催化剂对关键中间体OOH的吸附强度最强。由于ORR在Co-DABDT上的潜在决速步骤是O2加氢生成*OOH,因此Co-DABDT-2%对ORR具有最高的电催化活性。
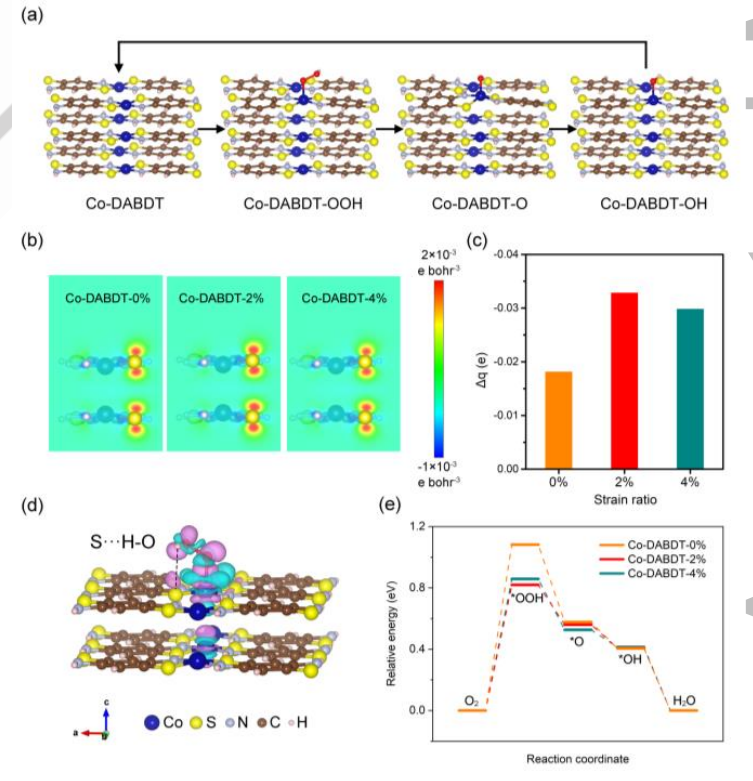
图4. 应变调节与电催化性能之间的DFT
研究总结展望
综上所述,本文成功地制备了具有特殊N、S配位性质的d-π共轭CoDABDT(DABDT=2,5-二氨基苯-1,4-二硫醇),且Co-N2S2在ORR过程中作为高金属活性中心。同时,通过利用溶剂体系的温差-压力特性,准确地调节了Co-DABDT的晶格应变,进一步提高了Co-DABDT的ORR活性。最优晶格应变Co-DABDT的ORR性能最为优异,其半波电位为0.81 V。此外,通过理论计算,深入说明了不同程度的晶格应变对Co-DABDT的电子结构和ORR性能的影响。晶格应变的存在主要影响Co-DABDT中S的自旋电荷密度(图1a),从而优化了中间体与催化剂之间的氢键相互作用,最终导致ORR过程的增强。这为理解电催化过程中金属配位框架的构效关系提供了一个新的视角。
文献信息Liu Lin+, Youxuan Ni+, Long Shang, Linyue Wang, Zhenhua Yan*, Qing Zhao, and Jun Chen*, Lattice Strained Induced Spin Regulation in Co-N/S Coordination Framework Enhanced Oxygen Reduction Reaction, Angew. Chem. Int. Ed.. (2024). https://doi.org/10.1002/anie.202319518